Toxicology Brief: Managing acute carprofen toxicosis in dogs and cats
Carprofen, a nonsteroidal anti-inflammatory drug of the propionic acid class, is commonly used in small-animal practice for its analgesic, anti-inflammatory, and antipyretic properties.
Carprofen, a nonsteroidal anti-inflammatory drug (NSAID) of the propionic acid class, is commonly used in small-animal practice for its analgesic, anti-inflammatory, and antipyretic properties. Its main uses in dogs are to treat pain and inflammation associated with osteoarthritis and control postoperative pain associated with soft tissue and orthopedic surgeries. More than 10 million dogs have been treated to date.1
Available as an injectable solution (50 mg/ml) and 25-, 75-, and 100-mg caplets and chewable tablets, Rimadyl (Pfizer Animal Health) is labeled in the United States for dogs only. The manufacturer's recommended dosage is 2 mg/lb (4.4 mg/kg) orally or subcutaneously daily (alternatively, 1 mg/lb [2.2 mg/kg] b.i.d.), and the injectable product is recommended to be given two hours before surgery to manage postoperative pain. Intramuscular and intravenous routes are extralabel and have also been reported.2 Generic formulations of carprofen—Vetprofen (Vétoquinol), Novox (Vedco), and Carprofen Caplets (Putney)—are also available as 25-, 75-, and 100-mg caplets for dogs only.3
The ASPCA Animal Poison Control Center (APCC) has received many calls regarding carprofen exposures in dogs and cats over the years (ASPCA APCC Database: Unpublished data, 2001-2009). This report details public cases in the computerized database (November 1, 2001 through April 27, 2009) that fulfill the following criteria in dogs and cats:
1. Single agent exposures
2. Observed exposures or those documented by evidence such as a chewed bottle
3. Clinical cases assessed by ASPCA APCC veterinarians as likely related to the agent
4. Acute overdoses only in dogs (to exclude idiosyncratic hepatic reactions)
5. Oral products only
THERAPEUTIC MECHANISM OF ACTION
Like most NSAIDs, carprofen is thought to mediate its beneficial therapeutic effects by inhibiting the enzyme cyclooxygenase (COX), which catalyzes the cyclization and oxygenation of arachidonic acid to prostaglandins. Discovered in 1991, COX-2 is the isoform of the enzyme induced by proinflammatory cytokines and mitogens,4 and COX-2 inhibition is the main intended target for the therapeutic effects of NSAIDs, particularly the more recently approved drugs. A higher ratio of COX-2 to COX-1 inhibition is associated with greater therapeutic efficacy and fewer adverse effects. However, in animals with underlying gastrointestinal disease or in those receiving concurrent NSAID or glucocorticoid therapy, any amount of COX-1 inhibition could be detrimental. Preexisting gastrointestinal inflammation, overdosage, and close temporal use of other NSAIDs or glucocorticoids in dogs receiving a selective COX-2 inhibitor (deracoxib) have resulted in gut perforation and death.5 The literature reports variable selectivity of COX-2 vs. COX-1 inhibition by carprofen.4,6-12
TOXIC MECHANISM OF ACTION
COX-1, the constitutive and cytoprotective isozyme, has many beneficial roles in the body. In the stomach, COX-1 reduces gastric acid secretion by parietal cells, maintains gastric mucosal blood flow mediated by vasodilation, and stimulates mucus and bicarbonate production by epithelial cells.13 Inhibiting this isozyme with NSAID therapy can result in gastrointestinal ulceration, hemorrhage, and gut perforation with septic peritonitis as a sequela. Direct damage to the gastric mucosal microcirculation and the formation of capillary microthrombi can also occur with NSAID use.13
In the kidneys, COX-1 activation results in prostaglandin I, E, and D production,14 which dilates renal vascular beds and diminishes vascular resistance, resulting in enhanced organ perfusion. Inhibition of these beneficial prostaglandins results in decreased renal blood flow, ischemia of the medullary papillae, and papillary necrosis.14
In platelets, COX-1 converts arachidonic acid to thromboxane A2, which is proaggregatory and vasoconstrictive. Its inhibition can be beneficial in preventing myocardial infarction in people3 and has been associated with subclinical increased bleeding times, decreased platelet aggregation, and decreased clot strength in dogs treated with a variety of NSAIDs.15,16
At therapeutic dosages, carprofen has greater selectivity for COX-2 vs. COX-1. Overdoses have been shown to increase COX-1 inhibition4 and the likelihood of adverse effects.
A subset of dogs treated with therapeutic doses of carprofen has exhibited an idiosyncratic hepatocellular toxicosis. A mean onset of clinical signs in dogs about 20 days after the start of therapy has been reported.17 Discontinuing the drug and administering supportive care resulted in complete recovery in most of the dogs in that report. This syndrome has not been reported in cats.
Because of the distinct pathologic mechanisms of idiosyncratic hepatocellular toxicosis vs. those of acute carprofen toxicosis, we will not discuss idiosyncratic hepatocellular toxicosis further. However, we will address hepatocellular injury due to intrinsic, dose-related effects associated with acute carprofen overdoses. The manufacturer reports about a 20-IU increase in alanine aminotransferase activity at doses 5.7 times the therapeutic dose in separate safety studies in dogs given carprofen orally for 13 weeks and one year12 and hypoalbuminemia in two of eight dogs treated at 10 times the therapeutic dose for 14 days.12
PHARMACOKINETICS
Pharmacokinetics plays an important role in carprofen toxicosis. Nearly 90% to 100% of the ingested drug is bioavailable,18 with a peak plasma concentration in one to three hours.2,19 Carprofen is also highly protein-bound, which can exacerbate the toxic effects of coadministered drugs that are also highly protein-bound, particularly drugs with a narrow margin of safety (e.g. anticoagulants, digoxin, methotrexate) as well as in dogs in which hepatic function may be abnormal.2,13 In dogs, the elimination half-life ranges from eight to 18 hours2,4,19; 70% to 80% of carprofen is metabolized by direct conjugation to an ester glucuronide followed by oxidation to phenol and further conjugation. These conjugated phenols are eliminated in the feces. Smaller amounts of carprofen are excreted as hydroxy metabolites in urine.2,20 Enterohepatic circulation has been documented for carprofen.19
Because of cats' limited ability to metabolize carprofen via glucuronidation, the drug's elimination half-life (20.1 ± 16.6 hours)21 in that species is longer than in dogs. Consequently, use of the drug in cats poses a far greater risk of adverse effects. In the United States, carprofen use in cats remains extralabel, so the pharmacokinetic differences in this species should be kept in mind, and extreme caution should be exercised when administering carprofen in cats.
ACUTE OVERDOSAGE
Because of dogs' relative lack of dietary discretion and the palatability of chewable Rimadyl tablets, it is not unusual for a dog to ingest an entire bottle of carprofen tablets if it is accessible (ASPCA APCC Database: Unpublished data, 2001-2009). With a maximum of 240 caplets per bottle and 100 mg per tablet,3 extreme ingestions are possible. And given the narrow margin of safety in cats, ingesting just one 25-mg tablet is potentially serious.
Table 1 lists the clinical signs reported by the ASPCA APCC in cases of acute overdoses in dogs. Vomiting is the most common finding, seen in 78% of cases, with an array of additional signs affecting the gastrointestinal system predominating thereafter. Fewer patients exhibit hepatic, renal, or neurologic signs or hematologic abnormalities. Nonspecific changes in energy status or mentation, such as lethargy or depression, are not uncommon.



TABLE 1: Clinical Signs Associated with Acute Carprofen Toxicosis in Dogs
Table 2 lists the comparable statistics for carprofen toxicoses in cats. With far fewer cases because of the relative dietary discretion of cats, the same array of signs can be seen with feline toxic ingestions.



TABLE 2: Clinical Signs Associated with Acute Carprofen Toxicosis in Cats
In otherwise healthy dogs, a review of the ASPCA APCC database historically indicated that fairly severe gastrointestinal signs may be seen at doses exceeding 20 mg/kg and that diuresis to prevent renal damage should be recommended at 40 mg/kg and above (ASPCA APCC Database: Unpublished data, 2001-2009).
Hepatic damage may occur with any dose, but the potential for nonidiosyncratic damage, by definition, should worsen with increased doses. According to the ASPCA APCC database, mild, transient, and often subclinical elevations of alanine transaminase (ALT) and alkaline phosphatase (ALP) activities have been seen with single, acute exposures beginning in the 40-mg/kg range in dogs. With single, acute exposures exceeding 100 mg/kg in dogs, more moderate to severe elevations in ALT and ALP activities can be seen, and these are often associated with clinical illness. Prophylactic administration of s-adenosylmethionine (SAMe) (17 to 20 mg/kg or more per day on an empty stomach) is recommended in dogs at greater risk for hepatic damage. A similar trend has not been identified in cats, but prophylactic SAMe administration (200 mg/kg on an empty stomach) should be considered in cats as well.
Neurologic signs, including seizures, stupor, and coma, tend to be seen with extreme ingestions. Respectively, these signs have been seen in ASPCA APCC cases in carprofen ingestions of 281, 536, and 645 mg/kg in dogs. Death despite treatment has been reported at 536 mg/kg. Definitive damage to the kidneys, as evidenced by the presence of urinary casts, was seen in a dog that ingested 48 mg/kg of carprofen.
In cats, the ASPCA APCC data indicate that ingestions of 4 mg/kg and above would be expected to cause more than mild gastrointestinal signs, and ingestions of 8 mg/kg and above may result in acute renal failure (ASPCA APCC Database: Unpublished data, 2001-2009). One 3-month-old kitten received 8.6 mg/kg of carprofen orally and was euthanized after acute renal failure developed. The lowest reported dose for death in a cat was ingestion of a possible range of 27.5 to 45.8 mg/kg (ASPCA APCC Database: Unpublished data, 2001-2009).
For both dogs and cats, keep in mind this information is from cases in which medical intervention had been sought and may not reflect the lowest doses at which signs may arise in untreated animals. Furthermore, because complete clinical findings and case outcomes are not always reported to the ASPCA APCC and because not all dosages are represented in the exposures reported, the lowest reported dosage at which a specific adverse effect has been seen may not be the true lowest dosage at which that effect is possible.
PRETREATMENT DIAGNOSTIC TESTS
Ideally, any animal with acute carprofen exposure expected to result in moderate or severe clinical signs should undergo pretreatment baseline diagnostic tests to better assess the patient's risk and to tailor the treatment. This assessment is especially true for carprofen exposure because a variety of underlying illnesses may complicate the toxicosis (see "Sidebar: Risk factors for carprofen toxicosis"). A complete blood count to obtain a baseline packed cell volume; a serum chemistry profile to assess blood urea nitrogen (BUN), creatinine, and electrolyte concentrations as well as liver enzyme activities; and a urinalysis to evaluate the urine specific gravity would be ideal.
TREATMENT
Treatment involves instituting decontamination, protecting the gastrointestinal tract and kidneys, providing supportive care, and monitoring gastrointestinal, renal, and hepatic function.
Decontamination
If the exposure history indicates a potential for adverse effects, decontamination is warranted. If a patient presents within a couple of hours of ingesting an overdose of carprofen and has no condition that precludes it, induce emesis. In dogs, administer 2.2 ml/kg of 3% hydrogen peroxide (maximum 45 ml) orally. You may repeat this dose if the patient fails to vomit within 15 minutes (ASPCA APCC Database: Unpublished data, 2001-2009). Alternatively, apomorphine can be given intravenously (0.04 mg/kg), subcutaneously (0.08 mg/kg), or in the conjunctival sac (0.25 mg/kg).2 Examining the vomitus often does not allow quantification of recovered medication because the residue of the chewable Rimadyl is often indistinguishable from partially digested food. Nevertheless, it is advisable to attempt to reevaluate the exposure by examining the vomitus.
Inducing emesis is an unpredictable endeavor in cats. The response to hydrogen peroxide is typically poor; apomorphine can stimulate the central nervous system, and xylazine (0.44 mg/kg intramuscularly) can cause central nervous system depression and increase the risk of aspiration.2 Decontamination with activated charcoal (e.g. ToxiBan—Vet-a-Mix) may be the best first step in cats.
After emesis has stopped, orally administer the first dose of activated charcoal. The ToxiBan label recommends 10 to 20 ml/kg for a small animal.22 Because of concerns for hypernatremia, however, the ASPCA APCC recommends a lower dose of 6.6 to 11 ml/kg of the 10% solution (alternatively, 1 to 2 g/kg of the 100% granules). A formulation containing a cathartic such as sorbitol is recommended for the first dose.
An experimental study in beagles involving an overdose of 16 mg/kg carprofen given orally documented the efficacy of administering 2.5 g/kg activated charcoal 30 minutes after exposure. The plasma carprofen concentration was significantly reduced with activated charcoal administration relative to the control group.23 Because of carprofen's enterohepatic circulation, multiple doses of activated charcoal can be beneficial. Subsequent doses without cathartic are given every eight hours and at a reduced dose (3.3 to 5.5 ml/kg of the 10% solution) to minimize the potential for osmotic diarrhea and hypernatremia. To further minimize the potential for these adverse effects, the number of doses of activated charcoal is typically limited to three, even with the most extreme overdoses.24
Gastrointestinal protection
Aggressive administration of gastrointestinal mucosa protectants (Table 3) is warranted as a precaution for seven to 10 days after exposure to carprofen overdoses (ASPCA APCC Database: Unpublished data, 2001-2009). The prostaglandin E1 analogue, misoprostol, is recommended for dogs, especially if the carprofen dose exceeds 20 mg/kg or the patient is exhibiting signs consistent with ulceration. Misoprostol is available in 100- and 200-μg tablets. Side effects include diarrhea, abdominal pain, vomiting, and flatulence, which may be confused with the underlying carprofen toxicosis. Because of the potential for adverse effects after several days of use, the veterinarians at the ASPCA APCC limit misoprostol administration to three to five days after exposure (ASPCA APCC Database: Unpublished data 2001-2009). Misoprostol is also used as an abortifacient, so its use is not recommended in pregnant dogs. Veterinary personnel or pet owners who may be concerned with this potential should wear gloves when administering the drug or avoid handling it altogether.2

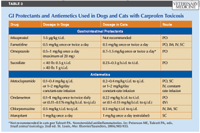
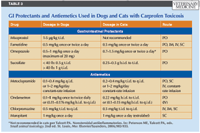
TABLE 3: GI Protectants and Antiemetics Used in Dogs and Cats with Carprofen Toxicosis
Additionally, gastric acid production can be decreased with an H2 antagonist, such as famotidine, or the proton pump inhibitor omeprazole.
A third measure to minimize the potential for ulcerative effects is to administer sucralfate, which reacts with hydrochloric acid in the stomach to form a protective paste at the site of ulceration.2 Sucralfate administration is recommended 30 minutes before an H2 antagonist or proton pump inhibitor since it requires an acidic environment to be efficacious.2
Prevent renal necrosis
To prevent renal papillary necrosis with carprofen overdoses, crystalloid fluid administration at twice the maintenance fluid rate (60 ml/lb/day) is indicated for 48 hours after exposure in dogs to promote diuresis. Carprofen's longer and more variable half-life in cats may necessitate diuresis for 72 hours. The choice of crystalloid should be dictated by a patient's electrolyte and acid-base status. If BUN and creatinine concentrations (see "Additional monitoring" below) are normal at 48 hours, the fluids may be tapered over 24 hours and then the renal values reevaluated at 72 hours. Monitor urine output throughout the diuresis period to ensure that the patient is not oliguric or anuric. Renal damage has been documented in one case as early as 24 hours after carprofen exposure by the presence of granular casts in the urine (ASPCA APCC Database: Unpublished data, 2001-2009).
ADDITIONAL SYMPTOMATIC TREATMENT
Antiemetics are indicated in actively vomiting patients (Table 3). If an animal is critically anemic, a whole blood transfusion is warranted. Severe hypoproteinemia or coagulopathy can be treated with a plasma transfusion (10 to 30 ml/kg intravenously).25 Vitamin K1 may be administered in patients with fulminant liver failure and coagulopathies.26 Diazepam may be given as needed for seizures. Pain due to gastrointestinal ulceration should be treated with opioid analgesia. Diuretics and dopamine may be necessary to restore urine production in oliguric or anuric animals. Hemodialysis27 or peritoneal dialysis may be an option in patients with acute renal failure if finances are not limited. A more thorough discussion of acute renal failure treatment is presented elsewhere.28-30 Hepatoprotective agents, such as S-adenosylmethionine (SAMe) or silymarin, can be administered in stable patients long-term until liver enzyme activities reveal no further hepatocellular damage. Broad-spectrum antibiotics are indicated if ulceration or hepatic compromise is present.
ADDITIONAL MONITORING
In patients hospitalized for diuresis, daily assessment of BUN and creatinine concentrations, packed cell volume, liver enzyme activities, and urine output and character (e.g. presence of casts, urine protein:creatinine ratio) and frequent monitoring of hydration status are indicated. Normal test results related to renal and hepatic function 72 hours after carprofen exposure are not anticipated to elevate thereafter. Pain due to gastric ulceration may not become clinically evident until the analgesic effect of carprofen has worn off, so monitor patients for several days thereafter for melena, anorexia, lethargy, and abdominal pain.
PROGNOSIS
If perforating or bleeding ulcers develop to the point of peritonitis or severe anemia, respectively, the prognosis understandably worsens. Severe azotemia, oliguria, anuria, and coagulopathy secondary to acute hepatic failure also carry a poorer prognosis. Even in extreme overdose cases, however, the prognosis with acute carprofen toxicosis is often excellent with prompt decontamination, diuresis, and gastrointestinal supportive care.
"Toxicology Brief" was contributed by Donna Mensching, DVM, MS, DABVT, DABT, and Petra Volmer, MS, DVM, DABVT, DABT, Department of Veterinary Biosciences, College of Veterinary Medicine, University of Illinois, Urbana, IL 61802. Dr. Volmer is the editor of "Toxicology Brief." Dr. Mensching's current address is ASPCA Animal Poison Control Center, 1717 S. Philo Road, Suite 36, Urbana, IL 61802. Dr. Volmer's current address is Summit VetPharm, 400 Kelby St., Fort Lee, NJ 07024.
REFERENCES
1. Managing pain with Rimadyl. Pfizer Animal Health Web site: http://www.rimadyl.com/display.asp?country=US&lang=EN&drug=RC&species=CN&sec=000. Accessed April 2009.
2. Plumb DC. Veterinary drug handbook. 6th ed. Stockholm, Wis: PharmaVet Inc, 2008.
3. Vétoquinol USA Inc: Vetprofen package insert. Fort Worth, Texas. http://www.vetprofenusa.com/pages/pro_vetprofen.html. Accessed April 2009.
4. Clark TP. The clinical pharmacology of cyclooxygenase-2-selective and dual inhibitors. Vet Clin North Am Small Anim Pract 2006;36(5):1061-1085, vii.
5. Lascelles BD, Blikslager AT, Fox SM, et al. Gastrointestinal tract perforation in dogs treated with a selective cyclooxygenase-2 inhibitor: 29 cases (2002-2003). J Am Vet Med Assoc 2005;227(7):1112-1117.
6. Brideau C, Van Staden C, Chan CC. In vitro effects of cyclooxygenase inhibitors in whole blood of horses, dogs, and cats. Am J Vet Res 2001;62(11):1755-1760.
7. Forsyth SF, Guilford WG, Haslett SJ, et al. Endoscopy of the gastroduodenal mucosa after carprofen, meloxicam and ketoprofen administration in dogs. J Small Anim Pract 1998;39(9):421-424.
8. Kay-Mugford T, Conlon P. Possible inconsistencies in study on cyclooxygenase. Am J Vet Res 1999;60(3):275-276.
9. Luna SP, Basilio AC, Steagall PV, et al. Evaluation of adverse effects of long-term oral administration of carprofen, etodolac, flunixin meglumine, ketoprofen, and meloxicam in dogs. Am J Vet Res 2007;68(3):258-264.
10. Ricketts AP, Lundy KM, Seibel SB. Evaluation of selective inhibition of canine cyclooxygenase 1 and 2 by carprofen and other nonsteroidal anti-inflammatory drugs. Am J Vet Res 1998;59(11):1441-1446.
11. Sessions JK, Reynolds LR, Budsberg SC. In vivo effects of carprofen, deracoxib, and etodolac on prostanoid production in blood, gastric mucosa, and synovial fluid in dogs with chronic osteoarthritis. Am J Vet Res 2005;66(5):812-817.
12. Pfizer Animal Health: Rimadyl Chewable Tablets package insert. Exton, Pa.
13. Talcott PA. Nonsteroidal antiinflammatories. In: Peterson ME, Talcott PA, eds. Small animal toxicology. 2nd ed. St. Louis, Mo: Elsevier/Saunders, 2006;902-933.
14. Roder JD. Analgesics. In: Plumlee KH, ed. Clinical veterinary toxicology. St. Louis, Mo: Mosby, 2004;282-284.
15. Brainard BM, Meredith CP, Callan MB, et al. Changes in platelet function, hemostasis, and prostaglandin expression after treatment with nonsteroidal anti-inflammatory drugs with various cyclooxygenase selectivities in dogs. Am J Vet Res 2007; 68(3):251-257.
16. Hickford FH, Barr SC, Erb HN. Effect of carprofen on hemostatic variables in dogs. Am J Vet Res 2001;62(10):1642-1646.
17. MacPhail CM, Lappin MR, Meyer DJ, et al. Hepatocellular toxicosis associated with administration of carprofen in 21 dogs. J Am Vet Med Assoc 1998;212(12):1895-1901.
18. Schmitt M, Guentert TW. Biopharmaceutical evaluation of carprofen following single intravenous, oral, and rectal doses in dogs. Biopharm Drug Dispos 1990;11(7):585-594.
19. Schmitt M, Guentert TW. Biopharmaceutical evaluation of carprofen following single intravenous, oral, and rectal doses in dogs. Biopharm Drug Dispos 1990;11(7):585-594.
20. Rubio F, Seawall S, Pocelinko R, et al. Metabolism of carprofen, a nonsteroid anti-inflammatory agent, in rats, dogs, and humans. J Pharm Sci 1980;69(11):1245-1253.
21. Parton K, Balmer TV, Boyle J, et al. The pharmacokinetics and effects of intravenously administered carprofen and salicylate on gastrointestinal mucosa and selected biochemical measurements in healthy cats. J Vet Pharmacol Ther 2000;23(2):73-79.
22. Vet-A-Mix, Inc: ToxiBan product labels. Shenandoah, Iowa.
23. Raekallio MR, Honkavaara JM, Säkkinan MS, et al. Effects of urine alkalization and activated charcoal on the pharmacokinetics of orally administered carprofen in dogs. Am J Vet Res 2007;68(4):423-427.
24. Gwaltney-Brant S, ASPCA Animal Poison Control Center, Urbana, Ill: Personal communication, July 2007.
25. Mathews KA, Scott H, Abrams-Ogg A. Transfusion of blood products. In: Mathews KA, ed. Veterinary emergency and critical care manual. Guelph, Ontario, Canada: Lifelearn Inc, 2006;667-681.
26. Schell L. Carprofen toxicity. Veterinary Information Network, updated 02/15/2006: http://www.vin.com/Members/Associate/Associate.plx?DiseaseId=307. Accessed April 2009.
27. Adin C. Dialysis: what do I need to know? in Proceedings. Western Vet Conf, 2004.
28. Labato MA. Strategies for management of acute renal failure. Vet Clin North Am Small Anim Pract 2001;31(6):1265-1287.
29. Cowgill LD, Francey T. Acute uremia. In: Ettinger SJ, Feldman EC, eds. Textbook of veterinary internal medicine. 6th ed. St. Louis, Mo: Elsevier/Saunders, 2005;1731-1751.
30. Ford RB, Mazzaferro EM. Kirk & Bistner's handbook of veterinary procedures and emergency treatment. 8th ed. St. Louis, Mo: Elsevier/Saunders, 2006;288-289.
















