Diagnosing and treating canine copper-associated hepatopathies
A review of the pathophysiology, clinical presentation, definitive diagnosis, and treatment
In the past 30 years, the impact of copper accumulation on hepatic function in dogs has received considerable attention. In some terrier breeds, an inherited metabolic defect compromises copper excretion; in other breeds, primary inflammatory hepatic disease may facilitate copper accumulation because of chronic cholestasis. Although substantial variability exists in patient presentation and the amount of copper accumulated, specific therapies aimed at reducing hepatic copper concentrations appear to be beneficial. In this article, we review the pathophysiology, clinical presentation, definitive diagnosis, and treatment of copper-associated hepatopathies in dogs.
COPPER METABOLISM
Copper is a trace element and an essential micronutrient. Adequate amounts are required as cofactors for numerous enzymes, electron transport proteins, and antioxidant molecules, all of which are crucial for everyday cellular function. After the stomach and small intestines absorb it, copper is transported into the portal venous circulation by the enterocyte via ATPase7A.1 Copper is rapidly taken up from the portal venous circulation by the liver, the principal organ involved in copper homeostasis.
Within the hepatocytes, copper proceeds through one of several metabolic pathways. It may be incorporated into enzymes for use within hepatocytes or into ceruloplasmin that is returned to the circulation for transport to extrahepatic tissues. Alternatively, copper may be stored within hepatocyte lysosomes as copper metallothionein or excreted by the hepatocyte into bile.1 Biliary excretion is the principal route by which unused or excess copper leaves the body.
COPPER ACCUMULATION AND HEPATOPATHY
Excessive ingestion, derangements in storage, or compromised excretion of copper leads to copper accumulation. Excessive hepatocellular copper accumulation overwhelms the lysosomal storage capacity, resulting in oxidative stress. Such stress leads to free radical formation, lipid peroxidation, and DNA damage. Hepatocellular damage leads to acute then chronic inflammation and, eventually, to cirrhosis.
In veterinary medicine, copper-associated hepatopathy was first described in Bedlington terriers in 1979.2 Since then, this breed-specific, autosomal recessive inherited disease has been well-characterized, and affected individuals are known to have inadequate biliary excretion of copper because of a gene deletion. This gene encodes a small cytosolic protein termed Murr1 that is required for the final process of vesicular copper movement and excretion at the canalicular membrane of hepatocytes.3 Affected Bedlington terriers can now be identified by genetic tests (CT-Marker, CT-Deletion—VetGen).
Copper has been implicated in several other breed-related hepatopathies, including disorders described in West Highland white terriers, Skye terriers, Dalmatians, Doberman pinchers, and Labrador retrievers. Findings in these breeds share some similarities with the Bedlington terrier disorder, but notable differences are evident. For example, progressive, lifelong copper accumulation has not been demonstrated in the non-Bedlington breeds. Also, the severity of liver damage does not necessarily correlate with hepatic copper concentrations; biopsy samples may show marked hepatitis with minimal copper accumulation. Since the primary mechanism for excreting unused copper is through bile, some researchers speculate that compromised bile flow is the underlying cause of copper retention in many of these other breeds.4 Cholestasis is a common finding in many hepatic disorders and could theoretically compromise biliary copper excretion enough to permit toxic accumulation.
Much uncertainty exists about the relevance of copper concentrations in liver samples with or without evidence of disease, and it may be necessary to develop breed-specific reference ranges. This controversy highlights the need for careful interpretation of laboratory data, biopsy results, and quantitative liver copper concentrations in light of a patient's breed and clinical condition.
CLINICAL PRESENTATION
Genetically predisposed Bedlington terriers accumulate hepatotoxic copper concentrations and manifest clinical signs by 2 to 4 years of age. Other breeds may develop clinical disease at any age depending on the severity and nature of their hepatic dysfunction.
Some dogs may present without clinical signs but with persistent blood work abnormalities, such as elevations in liver enzyme activities and other indicators of liver dysfunction. Dogs with chronic liver disease may present with weight loss, decreased appetite, polyuria and polydipsia, diarrhea, or intermittent vomiting. Jaundice, ascites, or hepatic encephalopathy may also occur in severely affected patients. Dogs may also present in acute crisis, which may reflect a sudden decompensation of occult chronic liver disease. Clinical signs may include anorexia, melena, vomiting, jaundice, or lethargy.
Rarely, severe acute hepatic necrosis may result in the release of stored copper into the blood. This release causes hemolysis, probably from direct oxidative damage to erythrocyte membranes. Affected dogs may present with pallor, jaundice, lethargy, inappetence, or pigmenturia. Hemolysis secondary to copper toxicosis has only been reported in Bedlington terriers.5,6
CLINICOPATHOLOGIC FINDINGS
The most consistent laboratory finding is increased alanine transaminase (ALT) activity. Other liver enzyme activities may be increased concurrently, including alkaline phosphatase (ALP), aspartate transaminase (AST), and gamma-
glutamyltransferase (GGT). The relative increase in ALT activity is often much higher than the relative increase in ALP activity, suggesting predominantly hepatocellular rather than cholestatic liver disease. Even mild increases in ALT activity are important and merit more attention than changes in ALP activity. In addition, consider nonhepatic causes of increased ALP activity, particularly if it is the only abnormal parameter.
Hyperbilirubinemia, hypoalbuminemia, hypoglycemia, low blood urea nitrogen (BUN) concentration, or hypocholesterolemia suggests considerable compromise to hepatic function. Elevated bile acid concentrations, ammonia tolerance testing results, or ammonia concentrations can confirm liver dysfunction and may provide prognostic information. However, they do not obviate the need for liver biopsy.
Since copper accumulation in the liver occurs slowly, substantial increase of ALT or AST activity in a previously healthy patient is most suggestive of an acute injury by a noncopper hepatotoxin. Hepatoprotectants, supportive care, and periodic monitoring (every two weeks) of serum chemistry profile results are indicated before liver biopsy is pursued.
Other laboratory abnormalities may include anemia from chronic disease (nonregenerative) or gastrointestinal blood loss (regenerative or nonregenerative). Liver disease predisposes patients to gastrointestinal ulceration because of impaired mucosal blood flow, which is a result of dehydration and portal hypertension and decreased clearance of histamine and gastrin.6 Evidence of gastrointestinal bleeding may include melena, hematochezia, or an increased serum BUN to creatinine ratio. Urinalysis results may reveal bilirubinuria, dilute urine (isosthenuria), or glucosuria.
ULTRASONOGRAPHY
Abdominal ultrasonography is indicated to rule out primary biliary tract disease, especially extrahepatic biliary obstruction, and can provide useful clues about the duration of a patient's liver disease. If the liver appears mottled (mixed echogenicity), small, cirrhotic, or nodular, the liver disease is probably chronic. If the liver appears unremarkable in terms of size and echogenicity in a patient with a one-time-only increase in liver enzyme activities, acute liver injury is more probable.

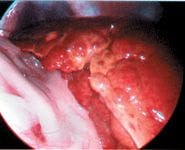
Figure 1. A laparoscopic photograph showing a nodular and discolored liver. (Photo courtesy of Dr. Mike Willard of Texas A&M University's College of Veterinary Medicine.)
DEFINITIVE DIAGNOSIS
Definitively diagnosing a copper-associated hepatopathy requires obtaining a liver biopsy sample surgically or laparoscopically (Figure 1) for histologic examination and copper quantification. Special stains (rubeanic acid, rhodanine, Timm's) can be used as a qualitative indicator of copper accumulation. Copper-loaded lysosomes can be identified with these stains when hepatic copper concentrations exceed 400 ppm on a dry weight basis (Figure 2).6


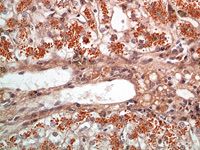
Figure 2. A photomicrograph from a liver biopsy demonstrating copper staining of copper-laden lysosomes (rhodanine stain, 400X). (Photo courtesy of Dr. Mike Willard of Texas A&M University's College of Veterinary Medicine.)
Atomic absorption analysis of liver tissue is the only way to accurately assess the hepatic copper concentration. Most laboratories require fresh or freshly frozen liver samples (Table 1). Copper concentrations are reported as µg/g of dry weight, which is the same as parts per million per dry weight (ppm dw).



Table1: Laboratories That Perform Heavy Metal Atomic Absorption Analysis
Copper concentrations > 2,000 ppm dw are thought to be directly hepatotoxic. However, some dogs may accumulate as much as 3,500 ppm dw before liver pathology is evident.1
In non-Bedlington breeds, copper concentrations are often substantially lower; Doberman pinschers with copper concentrations as low as 750 ppm may have morphologic evidence of hepatocellular damage and show improvement with copper chelation therapy.7 This supports the suggestion that the mechanism of copper accumulation may vary with different breeds.
Interpreting the histologic findings in these cases is an essential part of patient diagnosis. Important aspects to identify are the type (suppurative vs. lymphoplasmacytic) and extent of inflammation and the severity of necrosis, fibrosis (bridging is worse than piecemeal), and cholestasis.
TREATMENT
Only treatments for copper-associated hepatopathy are discussed below. In general, provide supportive care as required on a case-by-case basis, and discontinue any drug that is known to be potentially hepatotoxic (e.g. nonsteroidal anti-inflammatory drugs, phenobarbital). See Table 2 for therapy guidelines.



Table 2: Treating Canine Copper Hepatopathy*
Copper-restricted diet
Dogs with copper-associated hepatopathies should not be given soft water from copper pipes.6 Foodstuffs rich in copper, including shellfish, liver, kidney, heart, nuts, mushrooms, cereals, cocoa, and legumes, should also be avoided.6 In addition, these dogs should be fed a copper-restricted diet to slow—but not reverse—hepatic copper accumulation.
For growth and maintenance dog foods, the Association of American Feed Control Officials recommends 7.3 to 250 ppm per dry matter basis (DMB) of copper. Therapeutic veterinary diets designed for patients with liver dysfunction contain 3 to 5 ppm DMB of copper.6 Many of the therapeutic veterinary diets also contain high concentrations of antioxidants and adequate concentrations of high-quality proteins. Protein restriction is only required in dogs with hepatic encephalopathy, which is a rare complication.
Commercially available copper-restricted diets include Prescription Diet Canine l/d (Hill's Pet Nutrition) and Hepatic LS 14 Formula (Royal Canin). If a client prefers a homemade copper-restricted diet, consult a board-certified veterinary nutritionist.
Chelating agents
Copper chelators reduce liver copper content through chelation of copper in plasma and tissue, which is then excreted in the urine. D-penicillamine and trientine are two copper-chelating agents commonly used in veterinary medicine. D-penicillamine is associated with more side effects (e.g. vomiting, nausea, anorexia, lethargy, fever, skin problems) than trientine is, but it may also inhibit fibrosis by preventing cross-linking of collagen and by exerting an immunosuppressive effect by inhibiting T-lymphocyte function.8 Minimal, if any, side effects are associated with trientine use in dogs, but this product may be prohibitively expensive in large-breed dogs. When treating patients with D-penicillamine or trientine, it may take many months to remove excess copper from the liver (at an approximate rate of 900 µg/g dw per year) and for improvement in ALT activity to be appreciated.6,9
Copper chelation is not necessarily a benign treatment. One report in the veterinary literature described iatrogenic copper deficiency associated with long-term copper chelation in a Bedlington terrier.9 Clinical signs associated with copper deficiency can include central nervous system dysfunction, anemia, and abnormal ossification.5
Elemental zinc
Elemental zinc induces synthesis of intestinal mucosal metallothionein, which has a high affinity for copper and, thus, binds dietary copper and limits its absorption.1 The copper bound to the intestinal mucosal metallothionein is eventually excreted in feces as enterocytes are shed.1 Elemental zinc can be supplemented in the acetate, sulfate, gluconate, or methionine forms. Zinc gluconate is the most common formulation and is available over the counter. The dosage listed in Table 2 is for elemental zinc; clients may need help in selecting an appropriate product.
Zinc should be given one to two hours before a meal so the maximum amount of metallothionein will be available to bind copper. If nausea is reported, zinc may be given with a small amount of canned dog food. D-penicillamine and trientine could theoretically chelate zinc, but practically it is not a problem. Staggering the therapy should suffice.9
Ursodiol and glucocorticoids
Most of the information supporting the administration of ursodiol in patients with liver disease comes from studies in people. Ursodiol, or ursodeoxycholic acid, is a hydrophilic bile acid that shifts the bile acid profile toward the less toxic hydrophilic forms by competing with other bile acids for ileal absorption.8 In addition, the choleretic properties of ursodiol may increase copper excretion. Ursodiol may also reduce hepatocellular injury and fibrosis, modulate immune responses, and prevent bile acid-induced peroxidation by acting indirectly as an antioxidant.8
The goal of glucocorticoid therapy in patients with copper-associated hepatopathy is primarily to reduce inflammation. Since prednisolone is the active metabolite of prednisone and prednisone conversion may be impaired in dogs with substantial hepatic dysfunction, we prefer to use prednisolone in patients with liver disease. Prednisone and prednisolone have a 12- to 36-hour duration of action, and alternate-day dosing is ideal.8 Chronic corticosteroid use will increase ALP and GGT activities with minimal increases in hepatocellular leakage enzyme activities (ALT, AST); these changes do not reflect ongoing inflammation from copper-associated damage.
SAMe and vitamin E
S-adenosylmethionine (SAMe), a precursor of the antioxidant glutathione, is used as an antioxidant in liver disease. Also, it is recommended to help combat the oxidative effects of copper in copper-associated hepatopathy.8
Vitamin E is a potent antioxidant commonly used in treating liver disease, particularly copper-associated hepatopathies. Vitamin E is depleted in people with copper storage (Wilson's) disease, and its concentrations are decreased in chronic cholestasis.8 Advise owners to avoid giving their dogs selenium-containing preparations because of possible selenium toxicosis.
MONITORING AND MAINTENANCE THERAPY
Follow-up liver biopsies with copper quantification are required to determine efficacy and duration of chelation therapy. No consensus has been established about when it is appropriate to perform follow-up biopsies in these patients. In our experience, repeating the biopsy after 12 months of chelation therapy is helpful. The decision to perform subsequent biopsies can be based on serial monitoring of hepatic enzyme activities.
After effective chelation therapy, institute maintenance therapy to prevent copper reaccumulation. This therapy usually consists of zinc supplementation along with dietary copper restriction. Intermittent copper chelation therapy may be required to maintain normal hepatic copper concentrations in severely affected individuals.
CONCLUSION
Any patient with evidence of liver disease may have increased hepatic copper concentrations, either as a primary disorder or secondary to hepatocellular dysfunction and cholestasis. If a liver biopsy is performed for histologic analysis, submit a second sample for quantitative copper analysis—especially in known copper hepatopathy-associated breeds—since diagnosis and treatment may mitigate or reverse copper-associated changes.
Brier Bostrum, DVM
Audrey K. Cook, BVM&S, MRCVS, DACVIM, DECVIM-CA
Department of Small Animal Clinical Sciences
College of Veterinary Medicine and Biomedical Sciences
Texas A&M University
College Station, TX 77843
REFERENCES
1. Thornburg LP. A perspective on copper and liver disease in the dog. J Vet Diagn Invest 2000;12(2):101-110.
2. Twedt DC, Sternlieb I, Gilbertson SR. Clinical, morphologic, and chemical studies on copper toxicosis of Bedlington Terriers. J Am Vet Med Assoc 1979;175(3):269-275.
3. van De Sluis B, Rothuizen J, Pearson PL, et al. Identification of a new copper metabolism gene by positional cloning in a purebred dog population. Hum Mol Genet 2002;11(2):165-173.
4. Spee B, Arends B, van den Ingh TS, et al. Copper metabolism and oxidative stress in chronic inflammatory and cholestatic liver diseases in dogs. J Vet Intern Med 2006;20(5):1085-1092.
5. Rolfe DS, Twedt DC. Copper-associated hepatopathies in dogs. Vet Clin North Am Small Anim Pract 1995;25(2):399-417.
6. Guilford WG, Strombeck DR. In: Guilford WG, Center SA, Strombeck DR, et al., eds. Strombeck's small animal gastroenterology. 3rd ed. Philadelphia, Pa: WB Saunders Co, 1996;xiii,978.
7. Mandigers PJ, van den Ingh TS, Bode P, et al. Improvement in liver pathology after 4 months of D-penicillamine in 5 doberman pinschers with subclinical hepatitis. J Vet Intern Med 2005;19(1):40-43.
8. Sartor LL, Trepanier LA. Rational pharmacologic therapy of hepatobiliary disease in dogs and cats. Compend Contin Educ Pract Vet 2003;25:432-447.
9. Seguin MA, Bunch SE. Iatrogenic copper deficiency associated with long-term copper chelation for treatment of copper storage disease in a Bedlington Terrier. J Am Vet Med Assoc 2001;218(10):1593-1597, 1580.
10. Plumb DC. Plumb's veterinary drug handbook. 5th ed. Ames, Iowa: Blackwell Publishing, 2005;929.
















