NSAIDS and cats: what do we know? (Proceedings)
The cat as a species represents a therapeutic challenge when trying to use NSAIDs safey, including the newer drugs. Nonsteroidal anti-inflammatory drugs block the first step of prostaglandin synthesis by binding to and inhibiting cyclooxygenase This action is both dose and drug dependent.
The cat as a species represents a therapeutic challenge when trying to use NSAIDs safey, including the newer drugs. Nonsteroidal anti-inflammatory drugs block the first step of prostaglandin synthesis by binding to and inhibiting cyclooxygenase This action is both dose and drug dependent. The pharmacologic effects of this class of drugs and the relative dose associated with the effect include (antithrombotic, especially aspirin) <<< antipyresis < analgesia < control of inflammation. The role of PGs in normal physiology might best be understood by considering them as protective in nature. Their formation is mediated by one of at least two isoforms of cyclooxygenases. An inducible form (COX-2_ is measured following endotoxin stimulation (induction) of PG in macrophages. The constitutive form is characterized by baseline activity; designated COX-1, it is generally measured as thromboxane B2 synthesis from platelets. These or similar tests are used to generate a COX1:COX2 ratio which indicates the concentration of the NSAID that is needed to to inhibit 50% of the activity of either enzyme. A ratio > 1 indicates COX 2 is more easily inhibited than COX-1, often interepreted as a safer drug. However, these ratios must be studied in the species of interest and even then should serve only for screening purposes. Currently, one veterinary drug is approved for use for cats in the US (compared to 5 for dogs), although several others have been used effectively. What is not clear is which of these drugs prefer COX-2 to COX-1 in cats; to date, it appears that a ratio in cats has been reported only for carprofen (similar to dogs). NSAIDS with data addressing use specifically in cats include piroxicam, ketoprofen and carprofen. Tepoxalin (Zubrin®), the newest NSAID approved for use in dogs, is a "dual inhibitor", targeting both COX and LOX, thus targeting prostaglandins and leukotrienes.
Cox 1 versus Cox 2: Physiology and Pathology
In general, inhibition of COX-1 is responsible for efficacy whereas inhibition of COX-2 is responsible for side effects. However, strict adherence to this simplistic approach will lead to therapeutic failure and increased morbidity with NSAID use.
Central Nervous System:
In addition to pain, COX-2 has an important role in the pathogenesis of Alzheimer's Disease (AZD), and acute brain injury.
Pain
PGs have been implicated in causing increased pain perception (allodonia) in damaged compared to normal tissues. Induced COX-2 PGE as been associated with hyperalgesia (exaggerated response to pain) in either the spinal cord (primary hyperalgesia ) or at nociceptors in peripheral tissues (secondary hyperalgesia and central sensitization, manifested as a change in excitability threshold. Through COX-2, prostaglandins influence other CNS neurotransmitters (eg, glycine inhibition or glutamate stimulation) or other receptors (eg, NMDA). PGs also appear to mediate the anorexia and lethargy associated with chronic pain.
Gastrointestinal tract
Both COX-1 and COX-2 are constitutively expressed in the GI tract. Although COX-1 provides the major role in the protection of the GI tract, induction of COX-2 is important to healing of GI damage. Healing: The application of PGE-2 facilitates bone healing in experimental animals; 4 weeks of ibuprofen (16%) or rofecoxib (Vioxx®; 87%) compared to placebo (0%) led to mal-union in rats with experimentally-induced fractures.
Cardiovascular Disease
Platelets contain thromboxane synthetase, which catalyzes the formation of thromboxane from arachidonic acid. Thrombosis reflects platelet aggregation and vasoconstriction. The formation of a thrombus is kept "in check" by the presence of prostacyclin synthetase in vascular endothelial cells. This enzyme catalyzes metabolism of arachidonic acid to prostacyclin (PGI2), a vasodilatory and platelet inhibiting prostaglandin endproduct. However, whereas TXA2 is associated with COX-2, prostacyclin synthetase is associated with COX-1. The preferential inhibition of COX-2 may allow thrombus formation to go unchecked, increasing the risk of thromboembolic disorders. Thus, care must be taken when using these drugs in patients with hypertrophic cardiomyopathy.

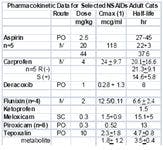
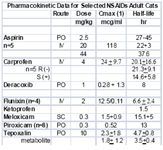
Kidney
In the kidney; both COX-1 and 2 are constitutively expressed. Both are formed in the macula densa of humans and animals, but COX-2 may have a more important role than COX-1. In (nonhuman) animals, inhibition of COX-2 causes sodium and potassium retention in salt depleted, but not normal, animals. However, in humans, COX-2 appears to influence renal vasculature and podocytes.
Lungs
The role of PGs in the lungs is less than that of leukotrienes; however, inflammatory diseases, such as asthma, are associated with smooth muscle proliferation which is inhibited by COX-2. Thus, like the GI tract, COX-2 appears to have a protective role in the diseased lung. Note that with older NSAIDs, inhibition of COX resulted in shunting of AA to LOX and increased production of leukotrienes. While this is not longer an issue in human medicine, the issue may remain in cats until the role of COX inhibition is clarified for each of the drugs. As such, NSAIDs should be used only with caution in cats with asthma. An exception might be tepoxalin.
Cancer
COX-2 increases markedly in a variety of soft-tissue tumors in humans and in transitional cell carcinoma in dogs (Knapp 2004). These studies suggest that benefits of NSAIDs in cancer may reflect inhibited COX-2. Depending on the model, inhibition of COX-2 associated with tumors reduces cell proliferation, increases apoptosis, and reduces metastasis. Inhibitors may also enhance anti-tumor effects of radiation, although toxicity is increased. However, at least for NSAIDs, GI toxicity is also increased when combined with antimetabolite anticancer drugs, presumably reflecting a combined toxic effect on the GI tract.
Disposition
Poor metabolizers have been described for some drugs in humans (eg. Asian descent) and dogs. It is tempting to speculate that breed differences will also emerge in cats of Asian descent. Adding to the complex impact of disposition on NSAID safety is the impact of enantiomers. Enantiomers are molecules that are mirror images of one another (R- or S+). They occur for many drugs which contain a chiral carbon around which its bonds rotate. Although chemically similar, the body handles each enantiomer differently in regards to disposition (see carprofen in table). Further, the pharmacodynamic effects of enantiomers are likely to be different. In essence, the body treats each enantiomer as a different drug. Most NSAIDs contain a chiral carbon, and are sold as racemic mixtures (50:50) of the R(-) or S(+) enantiomers. As such, extrapolation among species, genders, breeds, ages, etc becomes increasingly complex. Female cats already have been demonstrated to have a relative deficiency of CYP3A, the enzyme responsible for most drug metabolism.



Safety
Gastrointestinal damage is the most common and serious side effect of the NSAIDs among species. Cats are likely to be more sensitive to the GI side effects of NSAIDs compared to people. Because COX-2 is important to the healing ulcer, any NSAID should be discontinued at the first indication of GI upset. GI ulceration should be anticipated in an any animal receiving an NSAID. Treatment for GI toxicity should protect the damaged mucosa, and if necessary, control gastric acid secretion. Because (intestinal) ulceration is difficult without acid, the single most important treatment may be antisecretory drugs. In the face of severe ulceration, a proton pump blocker such as omeprazole may be most effective. However, its inhibitory effects on drug metabolizing should be avoided in patients receiving NSAIDs. Accordingly, famotidine (also can be given once daily and is minimally involved with drug interactions) can be used, especially from a preventative standpoint. As an inhibitor of hepatic drug metabolizing enzymes, omeprazole might be reserved for treatment of ulceration, when the NSAID is to be discontinued. The benefits of sucralfate include binding to and thus protecting damaged mucosa, as well as increased PGs synthesis, angiogenesis, and sulfhydryl (oxygen radical scavengers) production. However, sucralfate must bind to damaged tissue to be effective and thus is helpful in treating, but not preventing, ulceration.
Hemostasis
All traditional NSAIDs are able to impair platelet activity due to impaired prostaglandin (thromboxane) synthesis. At pharmacologic doses, aspirin selectively and irreversibly acetylates a serine residue of a platelet cyclooxgenase and, accordingly, will always have very low Cox 1: Cox2 ratios. However, newer NSAIDs inhibit prostacyclin, which acts to impair platelet aggregation, while minimally affecting thromboxane synthetase. As a result, thrombosis can occur relatively unchecked, predisposing patients to thrombosis (eg, Viox®). Unfortunately, it is unclear which of these drugs target predominantly Cox-2 in cats, decreasing confidence in use.
Renal
The cat may be predisposed to nephrotoxicity (compared to the dog or human), perhaps in part because of differences in renal function. In the kidney, vasodilatory PGs are protective, assuring that medullary vasodilation and urinary output continue during states of renal arterial vasoconstriction. The loss of this protective effect becomes important in patients with compromised renal function. Newer NSAIDs do not appear any less likely to be associated with this effect than the traditional NSAIDs in humans. In cats, a review of adverse drug events at the FDA's CVM website reveals that the proportion of adverse events of cats that reflect the kidneys (eg, 45% of meloxicam and 24% of carpfofen) is greater than that in dogs (9%), and is greater for meloxicam compared to carprofen. Unfortunately, this is the only comparative data available for NSAIDs and it has been removed from the website.
Drugs:
Meloxicam, like piroxicam, is a member of the oxicam group of NSAIDs The COX2:COX1 ratio for meloxicam, unlike that for piroxicam, favors selective COX2 inhibition in humans, suggesting that it has a wider margin of safety than most other. However, in cats, it is not clear if piroxicam is COX2 versus COX1-protective. Both piroxicam and meloxicam are characterized by a shorter half-life in cats compared to dogs. Meloxicam is more potent (although not necessarily more efficacious) than aspirin, indomethacin, and piroxicam; hence, its dose is smaller. The disposition of meloxicam has been studied in cats. Meloxicam is among the NSAIDs characterized by a shorter half-life in cats compared to dogs, and is one of the few NSAIDS that appear to be well tolerated in cats. It's safe use in Canada for several years predated its approval for use in cats in the US. Meloxicam is the only NSAID approved for use in cats in the United States. However, despite its apparent safety compared to other NSAIDs, the therapeutic margin of meloxicam is relatively narrow. Cats do not tolerate doses greater than or equal to 0.3 mg/kg gastric ulceration and death has occurred at 3X to 6X the normal for 10 days. Several studies support the efficacy of meloxicam in cats. In one study, the optimal dose of meloxicam to prevent endotoxin-induced fever in cats was 0.3 mg/kg, a dose also noted for its ability to cause toxicity. Meloxicam (0.2 mg/kg SC), carprofen (4 mg/kg SC), tolfenamic acid (4 mg/kg SC) and ketoprofen (2 mg/kg SC) did not differ in their ability to control post-operative pain following OHE in cats in one study. Meloxicam has recently been approved in Australia for chronic use at a dose of 0.05 mg/kg daily in cats. Genuw (J Fel Med Surg 2008) recently reported the safe used of meloxicam in adult (aged) cats using a case-control design (n=46; treated cats, age 12.9+4.2 yr) in Australia. Meloxicam was administered at 0.1 mg/cat (0.01-0.03 mg/kg (0.1 mg/cat) with food once daily following a 0.1 mg/kg once daily for 4 days oral loading dose. Mean duration of treatment was 5.8 months. However, there are some issues with the study. Since it was case controlled, no placebo of blinding took place and efficacy comparisons were based on comparison at baseline. Thus, it is difficult to assess efficacy. Illnesses in both the treatment and control group were diverse, with 25% of each group suffering from some disease. Ten pairs of cats were matched by disease. Cats with renal disease were included if disease was stable. One cat in each group died due to chronic renal insufficiency. Creatinine was measured only in the first 10 pairs of cats and only until 1 month of therapy. No statistical differences were found in creatinine between the treatment and control group (data not provided) at 1 month; however, the power of the study was not reported. One cat in each group died from chronic renal insufficiency; creatinine increased in cats with pre-existing disease treated with meloxicam numerically more than it did in control cats with the exception of 6 months (data not provided to evaluate variability). In addition, 4 cats in the meloxicam group and one in the control group developed vomiting. Caution is suggested when interpreting the results of this study as evidence of safety of meloxicam in regards to renal dysfunction associated with long term therapy. Caution is thus encouraged when using any NSAID, and perhaps meloxicam, in cats with pre-existing renal disease; this may include that population of cats whose serum creatinines are "high" normal.


Ketoprofen, although not firmly established, the efficacy of ketoprofen has also been attributed to its ability to inhibit some lipooxygenases and thus formation of leukotrienes. Ketoprofen is not approved for use in small animals in the United States but is approved for both dogs and cats in Europe. The half-life of the drug is short in cats (1.5 hrs) compared to dogs. Ketoprofen has been used as an analgesic in cats, particularly in Canada (1 mg/kg every 24 hours for 7 to 10 days). The antipyretic effect of ketoprofen (2 mg/kg subcutaneously followed by 1 mg/kg once daily orally) in febrile cats was rapid, being evident in 4 hours with temperatures normalized at that time. Temperatures did not change in the antibiotic-only treated cats. The use of ketoprofen as an analgesic is variable. In cats subjected to ovariohysterectomy, ketoprofen (2 mg/kg subcutaneously) compared favorably with buprenorphine (0.006 mg/kg or 6 mcg/kg intramuscularly) and mepiridine as gas anesthesia was discontinued. Response better for both drugs compared with the control at both 4 and 8 hours but still present for buprenorphine only compared with control at 18 hours. Carprofen is approved for use in cats in select countries outside the United States. The disposition of the drug has been studied in cats, including enantiomers at the dose associated with control of inflammation (4 mg/kg). A smaller clearance for carprofen in cats results in a elimination half-life that is at least twice as long in cats compared to dogs. The S isomer is cleared almost 3 times as rapidly as the R isomer resulting in a shorter half-life for S compared to R in the cat. The relative COX2 selectivity of carprofen that occurs in dogs has not been well documented in cats, although Brideau's data dose suggest relative selectivity similar to that in dogs. Clinically, however, this may not be true. The gastrointestinal effects of single dose carprofen (4 mg/kg IV) or aspirin (20 mg/kg IV) were studied in cats (n=5) using endoscopy and a randomized crossover design. Lesions in the stomach and duodenum 8 h postinjection were limited to minor pinpoint erosion in one cat. Clinical laboratory tests were not affected by either drug. However, duodenal perforation has been reported in cats using oral administration of carprofen (2.2 mg/kg twice daily for 7 days) following ovariohysterectomy. The ulceration was no doubt exacerbated by flunixin meglumine and dexamethasone treatment prior to referral. As a postoperative analgesic in cats, carprofen compares favorably with pethidine (meperidine), providing equal but longer analgesia (at least 24 hours) when administered a t 4 mg/kg subcutaneously postoperatively (Balmer et al., 1998). In a clinical trial of cats undergoing OHE, no difference was found in the analgesic effects of carprofen, ketoprofen, meloxicam or tolfenamic acid. based on the visual analogue scale and a nociceptive threshold at the incision site. With 9/10 cats responding, all responses were described as good, although none prevented wound tenderness.
Tepoxalin is among the dual acting NSAIDs. It is a potent anti-inflammatory and analgesic pyrazole derivative that also inhibits production of IL-1, suppreses NFkB activation and dependent gene expression (Keier 2004). Tepoxalin also has been studied in cats. Whereas tepoxalin was well tolerated in cats when administered at 100 mg/kg once daily for 3 consecutive days, saturation kinetics occur when 60 mg/kg was administered as two doses four hours apart. Signs suggestive of CNS ADE occurred (drunken-like state), a response not recorded in any other species. Tepoxalin is a potent antipyretic agent in cats at doses between 5 and 10 mg/kg and provides analgesia at least equivalent to butorphanol at 10 mg/kg for onychectomy (Personal Communication, Gerryll Hall, Technical Service Veterinarian, Schering Plough, April 2004).
Other drugs
Tramadol (Ultram) is a synthetic analogue of codeine currently marketed as a racemic (1:1) mixture of ± enantiomers. Tramadol appears to have multiple mechanisms of analgesia, with interaction among the pathways perhaps contributing to its efficacy. Opioid analgesia reflects agonistic interaction with mu receptors; additionally, tramadol enhances spinal pain inhibitory pathways through inhibition of neuronal re-uptake of serotonin (5-HT) and noradrenaline (NA), and release of 5-HT. As such, tramadol might be indicated for a broad array of conditions associated with pain, including chronic pain: studies suggest that the analgesia associated with 5-HT1A receptor agonists increases with chronic or repeat administration. Both tramadol stereoisomers are associated with analgesia, although receptor interactions appear to differ between the isomers. Tramadol is de-ethylated to an active metabolite, O-desmethyltramadol (ODT) which may be responsible for the majority of analgesic activity. The disposition of tramadol and ODT after either IV (2 mg/kg) or oral (5 mg/kg) administration of tramadol has been described in cats).The AUC of active metabolites is equal to or surpasses that of the AUC of the parent compound. The elimination half-life of both parent and ODT is approximately 2 fold higher in cats. Cats tolerated tramadol by either route, although they exhibited euphoria for several hours. A 5 mg/kg oral dose achieves for tramadol and well exceeds for ODT the minimum effective analgesic concentrations suggested in humans. It is not clear if a similar level of analgesia would be expected in cats, but based on the higher concentrations of ODT in cats, a dose of 2 mg/kg twice daily is a reasonable starting dose. Steagall studied the antinociceptive effects of tramadol (1 mg/kg SC) with or without acepromazine (0.1 mg/kg) in cats (n=8) subjected to thermal stimulation. Animals had only a limited response to tramadol, although the effect was increased when tramadol was combined with acepromazine. Care must be taken to NOT prescribe UltramPlus which contains acetaminophen. Gabapentin was initially developed as human anti-epileptic drugs. It is structurally related to GABA; gabapentin is a molecule of GABA covalently bound to a lipophilic cyclohexane ring. Although their mechanism of action was intended to be a GABA agonist, pharmacologically, they do not bind to an portion of the GABA receptor, nor do the appear to interfere with degradation or other aspects of GABA receptor activity. Rather, their mechanism of analgesic action appears to occur through binding to CaVa2-d proteins on voltage gated calcium channels. Decreased calcium influx prevents release of neurotransmitters otherwise stimulated by a variety of chemical signals. Gabapentin appears to be a safe anticonvulsant in cats and accordingly should be considered for long term control of pain.

















